|
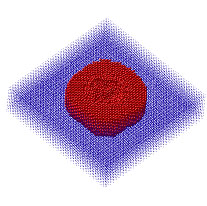
Extending the scope of atomic-scale simulations is a critically important goal because these types of simulations are required to fully understand the mechanisms responsible for the formation of many interesting nanoscale phenomena such as quantum dots, precipitate ordering, and mechanical fracture. Such features are currently beyond the reach of standard atomic scale simulations. In many situations of practical interest, not all the regions in a simulation are equally important, motivating the use of multiresolution approaches to reduce the computational requirements. Our research program in this area has been focused on the development of MD based simulations in which only a (changing) fraction of the atoms are actively considered, while atoms that lie far away from the feature(s) of interest are treated in a computationally expedient manner. An example of this approach, (which we call Feature Activated Molecular Dynamics, or FAMD) is given in Figure 1, which shows the evolution in an FAMD simulation of a spherical cavity under constant dynamic hydrostatic tension. The large red atoms surrounding the cavity are considered with standard MD, while the small blue ones further away are only subjected to occasional static relaxation. During the initial phases of the simulation, relatively few atoms need to be considered, and it is only in the final stages of the simulation, once the void has cavitated, that most of the atoms in the simulation cell are activated. The resulting computational savings are substantial and can be larger than a factor of ten over the entire simulation, depending on the size of the overall simulation cell.
Figure 1: Evolution of active atoms around a cavity subjected to hydrostatic tension. Initially only few atoms are activated and the simulation proceeds much more rapidly than a standard MD simulation. Towards the end, most of the atoms are active and standard MD speeds are achieved. Another example of this approach applied to a very different system is shown in Figure 2. Here a collection of about 2,000 point defects (self-interstitials) are introduced into a 1,000,000-atom silicon lattice and allowed to diffuse and agrgegate in time. Once again, only the regions of the lattice surrounding the defect clusters is of interest and the FAMD method tracks these regions as the clusters grow (or shrink) and diffuse within the lattice. Again, the computational savings relative to standard MD are substantial. This particular simulation was performed on a parallel computer platform with 8 processors using a parallel version of the FAMD algorithm.
Figure 2: Snapshots of an FAMD simulation of self-interstitial clustering in crystalline silicon. Total system size is 1,000,000 atoms but only red atoms are actually evolved with MD. Near end of simulation, only about 10% of the atoms are evolved with MD.
|





![]()